

Abstract
Genetic, epigenetic and transcriptional heterogeneity cooperate to fuel cancer's ability to evolve and adapt to therapy. In chronic lymphocytic leukemia (CLL), we have shown through bulk DNA methylation (DNAme) sequencing that the growing CLL populations diversify through stochastic DNAme changes (epimutations), impacting transcriptional heterogeneity, clonal evolution and clinical outcome.
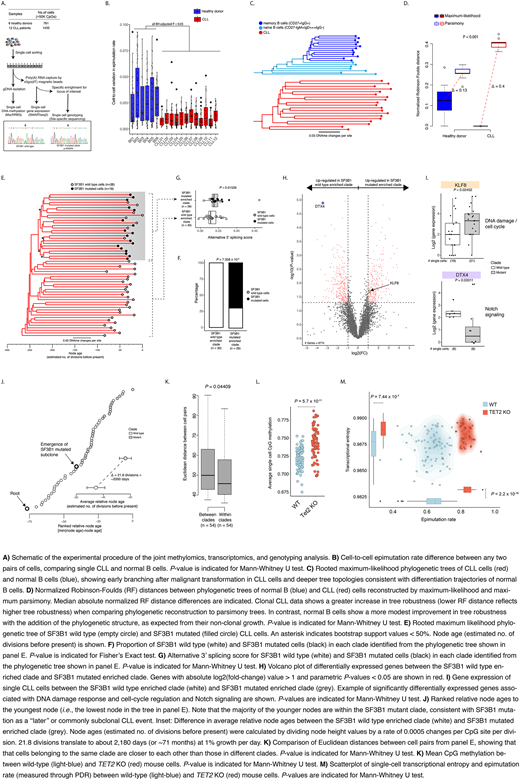
To directly integrate across epigenetic, genetic and transcriptional intra-leukemic cell-to-cell variation, we developed a high-throughput multi-modality platform that jointly interrogates the methylome, transcriptome and genetic driver mutations from the same single cell (Fig. A). We applied it to >2,000 B cells from 6 healthy donors and 12 CLL samples. We found that the common clonal CLL origin resulted in an elevated but uniform epimutation rate (i.e., low cell-to-cell epimutation variability). In contrast, in index sorted B cell subsets, ranging in maturity from naïve B cells (CD27-IgM+IgD+++IgG-) to memory B cells (CD27+IgG+), variable epimutation rates reflect cells with diverse evolutionary ages across the B cell differentiation trajectory (Fig. B).
Thus, we posited that epimutation can serve as a "molecular clock", enabling high-resolution lineage reconstruction, applicable directly to patient samples. CLL lineage tree topology revealed earlier branching, and longer branch lengths than normal B cells, consistent with rapid drift after transformation, and a greater proliferative history (Fig. C). In contrast to CLL topologies, non-clonal normal B cell trees provided a smaller increase in clustering accuracy compared with parsimony-based trees (P < 0.001; Fig. D).
To validate the inferred tree topology, we leveraged our multi-modal capture of DNAme and genetic drivers in single cells. Indeed, genetic subclones mapped accurately to distinct clades inferred solely based on epimutation information [e.g., a clade composed of SF3B1 mutants and another clade composed of SF3B1 wild-type cells (P = 7 x 10-9; Fig. E, F)]. Using the joint single-cell transcriptional profiling, we found that cells in SF3B1 mutated clade also displayed higher alternative 3' splicing than their wild-type counterparts (P = 0.01526; Fig. G). Cells belonging to SF3B1 mutated clade were marked by expression changes in genes related to DNA damage (e.g., KLF8) and Notch signaling (e.g., DTX4) (P < 0.05; Fig. H, I). The direct linking of single-cell transcriptional data with lineage identity also showed that transcriptional similarity between cells decreases as a function of their lineage distance (P < 0.05; Fig. K). Notably, the molecular clock feature of epimutations enabled precise timing of subclonal divergence event in the CLL's evolutionary history, estimated to have occurred 2180±219 days after the emergence of the parental clone (Fig. J).
To examine potential lineage biases during therapy, we performed serial multimodal single-cell profiling of a CLL patient without subclonal genetic drivers, prior to and during ibrutinib-associated lymphocytosis. The lineage trees revealed a distinct clade of cells preferentially expelled from the lymph node, marked by a distinct transcriptional profile, likely representing ibrutinib-sensitive cells.
Lastly, we hypothesized that frequent epigenetic modifier mutations seen in hematological malignancies may increase the epimutation rate promoting intra-leukemic cellular diversity. To test this hypothesis, we applied our multi-modality single-cell platform to cells from TET2 knock-out (KO) mouse models, and observed higher epimutation rates, closely associated with higher cell-to-cell transcriptional heterogeneity compared with wild-type cells (P < 0.0001; Fig. L, M).
In summary, we revealed that CLL show uniformly elevated epimutation rates, reflecting the common evolutionary age of these cells. By leveraging the heritable information captured through epimutation data, we provided a native lineage tracing applicable directly to patient samples. With this approach, we showed that CLL lineage topology exhibit early branching and long-branch length, consistent with exponential growth. Finally, we demonstrated that multimodal single-cell profiling enables projection of genetic and transcriptional identity onto the lineage tree, providing a direct measurement of the heritability of transcriptional profiles as a function of lineage distance.
Wu:Neon Therapeutics: Equity Ownership.

